1. Introduction
Fused Deposition Modeling (FDM) 3D printing is a technique for the manufacture of three-dimensional objects from the deposition of successive layers of a molten material of interest (sold in the form of cylindrical filaments with diameters of 1.75 mm or 3.00 mm) on a flat working surface of a 3D printer [
1,
2,
3]. Most applications in 3D printing involve the use of filaments based on thermoplastic polymeric materials, such as poly(lactic acid) (PLA), poly(ethylene terephthalate) (PET), and poly(acrylonitrile-butadiene-styrene) (ABS) [
4,
5,
6]. These polymers have been used in the production of conventional objects with different geometries, which meet demands in areas such as engineering, architecture and education.
The development of filaments based on biocompatible and biodegradable polymers is on the rise for applications in the field of pharmaceutical technology and biomedical engineering [
7,
8]. Emphasis is given to filament formulations based on poly(vinyl acetate) (PVA), poly(vinylpyrrolidone) (PVP), poly(ethylene glycol) (PEG), and poly(caprolactone) (PCL) (and their composites) containing drugs, intended for printing pharmaceutical solid solutions with personalized and reproducible geometries and dosages, for application in controlled drug delivery systems [
9,
10,
11,
12,
13]. Regarding the research and development of alternative and functional polymers for controlled drug release, Eudragit
® E 100 (E100) can be highlighted, whose solubility depends on the pH of the medium [
14]. It is a synthetic acrylic thermoplastic copolymer with good mechanical strength, and is biocompatible, biodegradable, and safe for human oral administration [
15].
This copolymer is already known as a polymeric matrix for protecting active substances and for masking the taste and odor of drugs, which facilitates their use when these characteristics become inconvenient for the user. Its cationic nature helps promote its rapid solubility only at low gastric pH (acidic, below 5.0). This intrinsic solubility is possible due to the hydration of the dimethylamino groups of its molecule, which causes these units to become protonated (acquire an H
+ proton), leading to the repulsion of these groups along the polymeric chain in continuous contact with the medium. With protonation, these functional groups interact with anions present in the gastric fluid to form salts, which drastically decreases the stability of the original molecule and breaks the copolymer chain [
16,
17]. On the other hand, in media with a basic pH, the copolymer molecule tends to become immobilized, and consequently the polymeric chain is impeded from flowing freely.
The E100 copolymer has been used in the pharmaceutical industry as a functional polymeric matrix (in homogeneous mixtures of the polymer with drugs) in pH-dependent pharmaceutical forms for oral administration, such as tablets, capsules, discs, and spheres. In addition, research over the last decade has explored the use of polymers in the form of films, ophthalmic solutions, sprays, and transdermal and vaginal implants [
18,
19,
20]. Its importance in programmed release formulations is also justified by the fact that this polymer can increase the dissolution rate and bioavailability of poorly water-soluble drugs, while improving the physical stability of amorphous solid dispersions [
14]. In this sense, the search for alternative dosage forms with E100, which are more efficient for encapsulation, transport, and controlled release of drugs in the gastrointestinal tract, is currently an open field of research in materials and pharmaceutical technology.
Although the importance of this copolymer is recognized in the industry as a coating and protection of active substances, and even in research for the development of micro/nanostructures for drug delivery [
21], works that deal with the development of filaments of E100 for 3D printing of customized pharmaceutical solid solutions are still incipient in the literature [
22,
23]. The main production challenges of this copolymer are linked to the the ability to manufacture filaments that are workable and functional, both during the filament molding process in extruder machines, and/or during the use in conventional 3D printers available on the market. The low flexibility and brittleness (rupture with low deformation), due to the cylindrical shape necessary for its use (high length compared to the diameter of the circular section), makes the continuous printing process of the designed pharmaceutical solid solution difficult.
Quality control of the E100-based filaments production using a hot-melt extrusion (HME) technique also involves thermal stability studies of the polymer in interaction with additives, since the process involves heating the material at high temperatures (generally, above 100 °C). It is known in the literature that the mass loss of the E100 influenced by temperature begins above 200 °C, with complete material degradation occurring above 300 °C [
24,
25,
26]. For example, temperatures in the range of 110 to 130 °C have previously been used for the development of E100/drug filaments containing various graft additives, using HME [
26]. In that invention, the temperature of 140 °C was also used in the printing process of E100 pharmaceutical solid solutions.
Sadia et al. studied the addition of varying amounts of tricalcium phosphate (Ca
3(PO
4)
2) and triethyl citrate (C
12H
20O
7) to E100, as an inert graft material and plasticizer, respectively [
22]. The authors reported that the optimized mass proportions of the materials were 46.75:3.25:50.00 of the copolymer, triethyl citrate, and tricalcium phosphate, respectively, for the preparation of filaments without drug addition, and of 46.75:3.25:37.50:12.50 of the same materials, for the preparation of filaments with the addition of 12.5 wt% of the drug. In the study conducted by Choudhury, Murty, and Banerjee [
23], considerable amounts of magnesium silicate (MgSiO
3) and triethyl citrate were used as additives. Triethyl citrate was used for the same purpose, as reported in the work by Sadia et al. [
22]. Magnesium silicate was used as an anti-adherent agent so that the copolymer would not adhere to the walls of the extruder tube during the hot melt extrusion process. With the presence of these additives, the mass of active principle per volume of filament (in mg/cm
3) was also automatically reduced.
In this context, this work aims at the preparation and characterization of 3D printing cylindrical filaments of the Eudragit E100 copolymer, containing variable amounts of the Hesperidin (Hsp) model drug (flavonoid, with antioxidant and anti-inflammatory properties), and respective in vitro controlled release tests of the active molecule, from the E100 matrix. The E100/Hsp hybrid filaments were produced using hot-melt extrusion (HME), and then they were subjected to a surface chemical protonation treatment, free of chemical additives. The microstructural characterization of the filaments was performed using SEM, XRD, FTIR, and ATR techniques, while the thermal analysis of the materials was performed using DSC. The mechanical behavior of the samples was evaluated from flexion tests in a universal testing machine. The in vitro Hsp controlled release tests were carried out in simulated gastro and intestinal solutions, with the aim of obtaining the temporal variation in the relative concentration of Hsp released in these fluids. The results showed filaments with uniform diameter which were able to preserve the polymer and the drug molecular structures after the extrusion process. In addition, it was noted that the protonation process was able to improve the flexibility of the E100 filaments, compared to those produced without chemical treatment.
2. Materials and Methods
2.1. Materials
Eudragit® E100 copolymer (E100; granules; Evonik Industries, Essen, Germany), Hesperidin (Hsp) (C28H34O15; powder; Sigma Aldrich, St. Louis, MI, USA), monobasic potassium phosphate (KH2PO4; powder; Sigma Aldrich, St. Louis, MI, USA) and sodium hydroxide (NaOH; 1N solution; Neon, Sao Paulo, Brazil) were used as received.
2.2. Sample Preparation for the Extrusion Process
The samples of E100/Hsp hybrid filaments for the hot-melt extrusion process were prepared by mechanically mixing varying amounts of Hsp (low level (80 mg) and high level (240 mg), so-called E100/Hsp(−) and E100/Hsp(+), respectively) with 24 g of the pH-dependent polymer E100, without additive reagents, until complete homogenization of each mixture (
Figure 1). Pristine E100 was also used in the experiments as a control sample for result comparison purposes.
2.3. Filament Production
The filaments were produced using a hot-melt extrusion process, followed by a surface protonation process of the resulting material. For the extrusion process, a mini-extruder machine (Filastruder
®, Snellville, GA, USA) (
Figure 2a) with the following characteristics was used: cylindrical metal reservoir measuring 25 cm in length and with an internal diameter of 1.5 cm (
Figure 2b,c); galvanized conveyor spindle; 1.75 mm extruder nozzle; extrusion speed of 40 cm min
−1; and vertical position (
Figure 2d).
The extruder received adaptations to meet the specificities of the E100 polymer. The reservoir (which holds the materials for extrusion) was initially divided into two parts (
Figure 2b) to facilitate the complete cleaning of the inner walls in contact with the E100/drug mixtures after each experiment. The tube parts received flanges at one end, allowing for the union of the parts with the fixation of screws (
Figure 2c).
The temperature of the extrusion system was set at 130 °C and held unchanged for 1 h. Then, each prepared mixture was gradually deposited inside the heated metal reservoir and kept at that same temperature for 10 min, so that the heat transfer from the tube to the material occurred uniformly.
Afterwards, the spindle that transports the materials inside the tube was turned on and the extrusion of cylindrical filaments was carried through the extruder nozzle. The produced filaments were collected and stored in circular plastic spools (15 cm diameter and 25 mm thickness). For the protonation chemical process of the E100 polymer chains, the filaments arranged on the circular spools were placed in a closed cubic plastic box with dimensions 30 × 40 × 30 cm3, connected to the output of a conventional air humidifier. In the box, each filament received from the device the vapor (60% relative humidity) of a solution prepared with 6.8 g of potassium phosphate and 77 mL of 0.2 M sodium hydroxide, in 1000 mL of deionized water (pH = 6.5 ± 0.1), for 60 min. After the chemical treatment, the samples were dried at room temperature for 24 h before starting the tests.
2.4. Microstructural Characterization
The materials microstructure was analyzed using (i) Scanning Electron Microscopy (SEM) (Vega 3XM Tescan apparatus, equipped with energy dispersive X-ray detector (EDX), accelerating voltage of 10–20 kV). For the experiments, the samples were covered by sputtering with a gold layer (Q150R ES Quorum® device), with a 5 nm min−1 rate, for 5 min; (ii) Fourier Transform Infrared Spectroscopy (FTIR, Shimadzu Prestige 21 equipment, in the wavenumber range from 4000 to 600 cm−1), using KBr method (for the evaluation, pressed pellets formed from a homogeneous mixture of 10 mg of each macerated filament with 1 g of KBr crystals were used) and attenuated total reflectance (ATR) methodology (Shimadzu apparatus, 4000 to 500 cm−1 range); and (iii) X-ray diffraction (XRD, Miniflex Rigaku equipment, Cu Kα radiation, λ = 1.54056 Å, 40 kV voltage, current of 15 mA and scanning rate of 0.02°s−1 in the 2θ range of 5° to 50°). X-Pert HighScore software v. 5.2 was used to identify the phases of the materials.
2.5. Thermal Analysis
The thermal behavior of the materials was analyzed using Differential Scanning Calorimetry (DSC), using a DSC-60 Plus device (Shimadzu, Kyoto, Japan). A mass of 2 mg of the produced filaments was placed in circular aluminum pans with lids (diameter of 2.5 mm, Shimadzu, Kyoto, Japan), and analyzed in the range of 30–285 °C, with a heating rate of 10 °C min−1.
2.6. Flexural Testing
The mechanical resistance testing of the filaments to flexion efforts was carried out in an EMIC DL 10,000 Universal Testing Machine (EMIC, São José dos Pinhais, PR, Brazil), equipped with a 500 N-capacity load cell. The experiments were conducted in triplicate, using cylindrical filaments as test specimens. The distance between the support points of the specimens used in the tests was 40 mm and the speed of the load cell for evaluating the samples was set to 10 mm min−1, in accordance with the ASTM D790 standard for semirigid/rigid rod-like polymeric materials. The stress (σ) and strain (ε) data obtained in the tests were collected using TESC® 3.04 software and examined to determine the rupture time of the samples and their respective modulus of elasticity (Young’s Modulus), E = tg(α) = ∆σ/∆ε, where α is the angular coefficient in the linearity region (elastic zone) of the σ versus ε plots.
2.7. In Vitro Dissolution Studies
The in vitro release profiles of the drug-containing filaments were evaluated in solutions of simulated gastro and intestinal fluids (at pH 1.2 and 7.2, respectively), in accordance with the procedure established by the United States Pharmacopeia National Formulary (USP-NF-Reagents) [
27,
28,
29]. Thus, 100 mL of each solution was disposed in 250 mL beakers and kept in a water bath at 37 °C, with a constant stirring of 100 rpm [
27,
28,
29].
Afterwards, samples of each composite filament were immersed in the solutions for analysis of the temporal variation in drug concentration in the media (in triplicates,
Figure 3a). The experimental apparatus used in the dissolution tests is shown in
Figure 3b.
Then, 2 mL aliquots of each solution were removed from the beaker at fixed time intervals to determine the intensity variations in Hesperidin absorption peak (λ= 283) in a Kasvi K-37 UV–vis spectrophotometer (Kasvi, São José dos Pinhais, PR, Brazil). The absorbance data were converted into relative concentration of the drug in solution ((Ct − C0)/C0, where C0 and Ct represent the initial and time t concentrations, respectively) from the respective calibration curve.
2.8. Statistical Analysis
The calculated filament diameter is the average (± standard deviation) of three independent measurements, for each sample configuration. These values were obtained from SEM micrographs, using the ImageJ software v. 1.53t. Inferential statistical analyses (normality and equality of means tests) were performed with a level of significance (α) of 0.05 and compared with the p-value obtained using these tests. For p-value > α, the hypotheses of normality and equality of means are accepted.
3. Results and Discussion
The stress (σ) versus strain (ε) plots for the filament specimens, without and with chemical treatment, are shown in
Figure 4. The graphs (i), (ii), and (iii) represent the results for pristine E100, E100/Hsp(−), and E100/Hsp(+) filaments, respectively, without chemical treatment. These samples demonstrated similar characteristics, with high rigidity and fragility to the normal force applied to their surfaces, confirmed by the values of the elasticity modulus (E = ∆σ/∆ε, in the elastic zones of the curves—dashed lines) estimated for 210, 189, and 380 MPa, and by the specimen rupture time of 41.7 s, 39.1 s, and 21.0 s, respectively (from sample (i) to (iii)).
This behavior is typical of the combination between the acrylic nature of the E100 copolymer and its cylindrical volumetric geometry (with a high length/circular section area ratio) which is necessary for its use as a filament. In addition, the samples of hybrid filaments E100/Hsp(−) and E100/Hsp(+) that were chemically treated (samples (iv) and (v), respectively), no longer presented the characteristics of pure elastic structures, with a significant reduction in E for 75 and 130 MPa (a 60% reduction for E100/Hsp(−) and 65% reduction for E100/Hsp(+), in comparison to the initial samples), and with rupture times of 116.6 s and 101.1 s, respectively, which explains the significant increase in the filaments’ flexibility.
Under the action of steam from the protonation solution (pH = 6.5, slightly acidic, close to the basic medium transition), the dimethylamino groups of the polymeric molecule are repelled from each other, from the outermost layers of the filament to the bulk of the material (with a radial direction,
Figure 5a). When the process is interrupted, the dimethylamino functional group in the more superficial monomers tends to retract, making it difficult for this event to occur in the inner layers of the filament. This gradient gives the filament the highest flexural strength noted in the tests. An image of the flexible filaments is shown in
Figure 5b. In addition, it was noted that 24 g of polymer, in each experiment, can produce around 4 m of filament with constant diameter, for each of the three types of materials tested (E100, E100/Hsp(−) and E100/Hsp(+)).
Figure 5c exemplifies a sample of continuous and complete E100 filament obtained via hot-melt extrusion (HME), at 130 °C.
The model drug Hesperidin (Hsp, as obtained from the supplier), and the produced flexible filaments E100, E100/Hsp(−) and E100/Hsp(+) were analyzed using SEM (
Figure 6). Hsp appears as a particulate material, with irregular geometry, and heterogeneous particle size distribution in the range of 2–20 µm.
The pristine E100 filament (control sample) presented a regular surface, without apparent structural defects (such as holes and large variations in diameter) that could negatively interfere with its usability (
Figure 6b). These filaments showed a normal distribution of diameters, with a mean value of 1.78 ± 0.12 mm (
n = 25). In addition, the E100/Hsp(−) filaments (
Figure 6c) and E100/Hsp(+) (
Figure 6d) showed surface characteristics similar to the E100 sample, with a mean diameter of 1.79 ± 0.18 mm (
n = 33) and 1.81 ± 0.13 mm (
n = 28), respectively. These results also indicate that the extrusion temperature for the production of E100/Hsp filaments was adequate, since excess heat during extrusion could cause the appearance of irreversible bubbles in the filaments during their solidification and, consequently, result in geometry discontinuities.
The equality of means hypothesis test demonstrated that there is no significant difference in the average diameter of the cross sections of the produced filaments (95% confidence level), with or without drug addition, both for low or high levels of available drug mass. This is an indication that these filaments do not show significant changes in volume per unit length, after the introduction of variable amounts of the drug. This last analysis is important for the purpose of the research because it demonstrates that the filaments produced with or without additives do not show significant variation in the surface area in contact with simulated gastrointestinal fluid solutions for in vitro release tests.
Thus, the temporal variation in drug concentrations released in these media (initially available in the volume of the filament) will be directly related to the pH-dependent nature of the polymer and with the mass of drug available in the bulk of the filament (proportional to its length). This proportionality and the homogeneous disposition of the drugs in the filaments are important factors for the production of pharmaceutical solid solutions printed with different geometries and replicable formulations [
30].
Figure 7 presents the X-ray diffractograms (XRD) of the filament samples and of the drug Hesperidin (Hsp). The diffractogram for the pristine E100 filament type exhibits the usual amorphous characteristic of the copolymer in its original form, with a broad peak in the 2θ range of 12–27° [
31,
32]. On the other hand, the spectrum of the pure Hsp demonstrates the typical crystalline nature of the drug, with narrow and intense peaks at specific 2θ in the analyzed range (at 12.4°, 13.8°, 15.7°, 16.4°, 19.8°, 21.4°, 22.5°, and 24.9°) [
33,
34,
35].
The XRD spectrum of the E100/Hsp(−) filament sample shows the amorphous predominance of the polymer, as previously discussed. The specific diffractogram demonstrates that the characteristic peaks of the crystalline phase of Hsp do not appear explicitly in the spectrum, due to the use of a small proportion of the drug in comparation to the mass of E100 in this sample and/or the appearance of the amorphous phase of the Hsp.
In addition, detailed zoomed portions of
Figure 7, (1) and (2), show the existence of two diffraction peaks of the drug in the E100/Hsp(−) filaments, at angles 2θ = 12.4° and 21.4°, indicating the presence of Hsp in the composite. The XRD spectrum of the E100/Hsp(+) filament has its baseline shifted to higher intensities, following the curvature of the E100 spectrum, in the range 2θ = 12–27°. In this same range, there are peaks that were previously detected in the Hsp spectrum, which indicates the presence of the crystalline phase of the drug in the filament.
These results represent a first indication that the E100 pH-dependent characteristic and the pharmacological properties of Hsp are maintained by the filaments after the hot-melt extrusion process.
FTIR spectroscopy was used as a tool for detecting molecular vibrations in the materials used in the filaments production. The spectra are presented in
Figure 8. The FTIR spectrum of the filament produced with pure E100 (E100 sample) exhibits characteristic vibration bands of the polymer (provided by the manufacturer) at 2954 cm
−1, 2770 cm
−1, 2873 cm
−1, 2820 cm
−1, 1729 cm
−1, 1460 cm
−1, 1385 cm
−1, and 1150 cm
−1. These vibrational bands are in agreement with results obtained in previous works in the literature (as detailed in
Table 1).
The FTIR spectrum of the Hsp also returned all characteristic vibration bands of the drug, at 3545 cm
−1, 3477 cm
−1, 2980 cm
−1, 2918 cm
−1, 1647 cm
−1, 1519 cm
−1, 1444 cm
−1, 1300 cm
−1, and 1276 cm
−1. These vibrational bands are detailed in
Table 2. The spectra obtained for the E100/Hsp-type filament samples were similar. In addition, the hybrid filaments present the stretching and bending vibrations of the separated materials before extrusion, indicating the viability of the production process for these composites, with combined characteristics of the polymer and the drug.
In addition to the FTIR data, the attenuated total reflectance (ATR) method was used to verify the solubility of E100, E100/Hsp(−), and E100/Hsp(+) filaments when immersed in basic pH fluid (inset of
Figure 8). The results of the analysis of the solution where the samples were immersed, after 60 min under constant agitation, showed the total absence of vibrational bands of the polymer and drug, across the entire wavenumber range studied (with negligible variation in transmittance intensity from 500 to 4000 cm
−1). In fact, the E100 polymer is highly hydrophobic and this characteristic is already widely known in the literature for the synthesis of nanostructures to encapsulate drugs [
17]. When immersed in fluids with basic pH, the E100-based filaments retain their chemical structure and their shape remains unchanged, without gaining mass and/or volume.
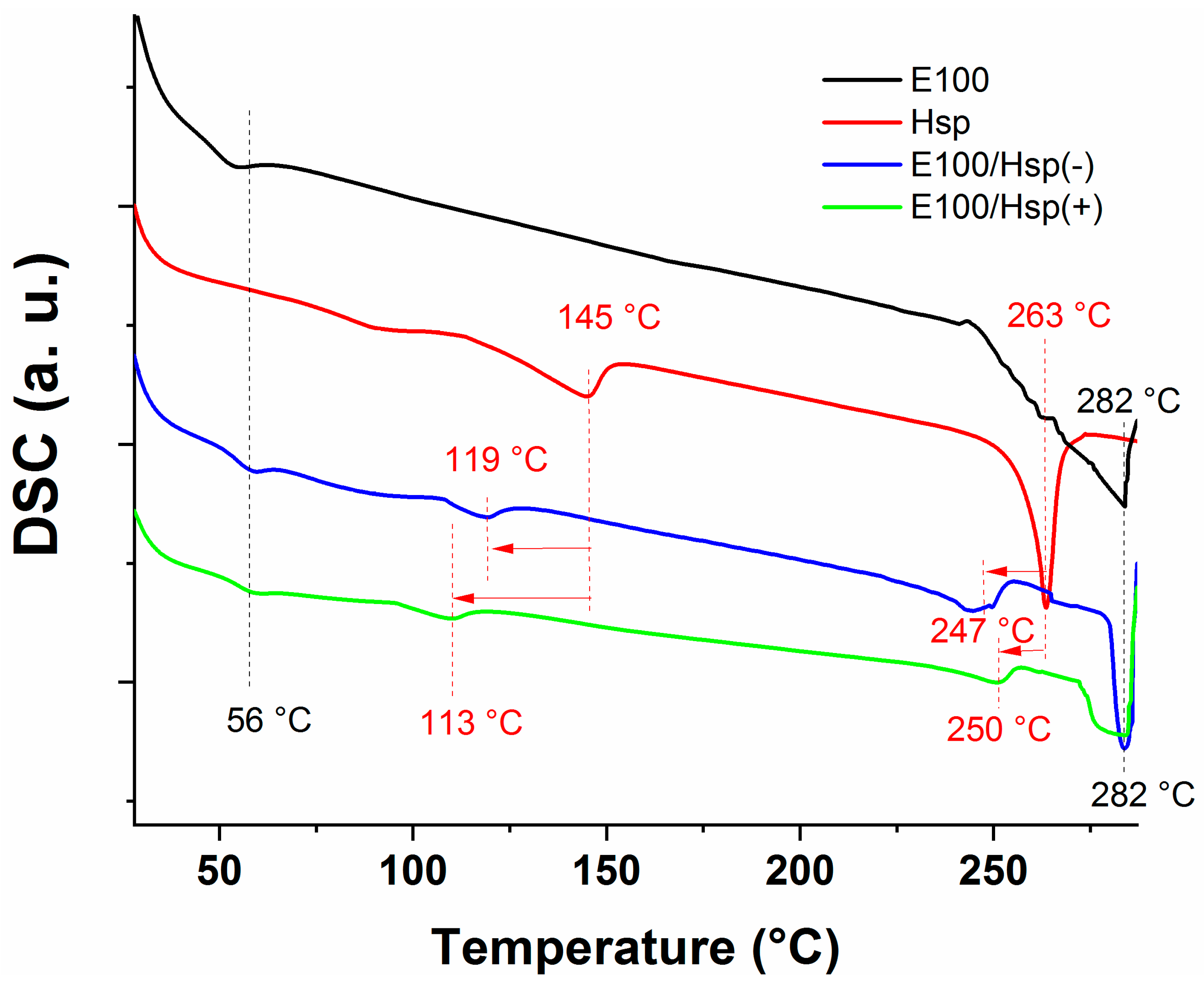
The thermal analysis of the E100, E100/Hsp(−) e E100/Hsp(+) filaments and pristine Hsp were investigated based on the obtained DSC spectra (
Figure 9). The E100, E100/Hsp(−), and E100/Hsp(+) DSC spectra of the filament samples display an endothermic peak at 56 °C, which corresponds to the glass transition point of the polymer [
17,
26,
42]. In addition, the signal noted at 282 °C (for all three filament types) corresponds to the degradation temperature of the E100 [
17].
The DSC spectrum of Hesperidin (Hsp) showed an endothermic peak at 263 °C, corresponding to the melting point of the drug, in agreement with data in the literature [
43,
44,
45]. No signal of degradation of the Hsp molecule was detected in the temperature range analyzed, which endorses the thermal stability of the compound at the processing temperatures of the filaments. The DSC thermogram of pure Hsp also shows a smaller peak centered at 145 °C (not widely discussed in the literature), possibly due to its glass transition temperature occurring above 100 °C or to a dehydration process in the material [
35,
46]. This temperature is shifted to 119 °C and 113 °C for the E100/(Hsp(-) and E100/(Hsp(+) filaments, respectively. These changes do not influence the usability of the drug for its intended purpose.
The DSC spectra also highlighted that the melting point temperature of the Hsp molecules in the E100/(Hsp(-) and E100/(Hsp(+) filaments was shifted to lower temperatures (247 and 250 °C, respectively), with a lower intensity of the respective endothermic peaks.
These lower intensity peaks suggest that the Hsp available in the filaments is dispersed in the amorphous matrix of the polymer, with an increase in the drug contact surface area. This dispersion is an important result for the application of the filaments containing Hesperidin, considering that the crystalline phase of the drug is poorly soluble in water and has a low dissolution rate when administered in the form of oral solid solutions (a limiting factor for its bioavailability).
Thus, as the filaments were extruded at 130 °C (a temperature much lower than the Hsp melting temperature), part of the Hesperidin micrometric particle agglomerates (as observed in the SEM results) tends to dissolve and/or to decrease in size in the polymer bulk during extrusion. This behavior, already mentioned for other Hesperidin solid dispersions using poly(ethylene glycol) and poly(vinyl pyrrolidone) as the polymer matrix, facilitates the drug dissolution and absorption in the action medium [
45,
47].
Figure 10 shows the DSC curves of the E100, E100/Hsp(−), and E100/Hsp(+) filaments and of E100 granules (as provided by the manufacturer), in the range 30 to 135 °C, for two complete heating and cooling cycles. It is possible to observe that all filament samples (materials that have previously been heated to 130 °C) present thermal characteristics similar to those of the E100 polymer in its original form, during the two test cycles, during which no degradation of the materials is detected. In fact, it is known from the literature that the degradation of the E100 polymer begins above 200 °C, and that complete degradation occurs above 300 °C [
24,
25,
26] (not only is the thermal stability of the polymer at this extrusion temperature shown, but also that there is no loss of material mass up to around 200 °C).
Thus, it is possible to infer from the combined analysis of the DSC, XRD, FTIR, and ATR spectra that the hot-melt extrusion (HME) process at 130 °C is suitable for the production of E100/Hsp hybrid filaments for biomedical use, since the process does not cause changes in the chemical composition and/or the degradation of the polymer or the drug [
24,
26,
42].
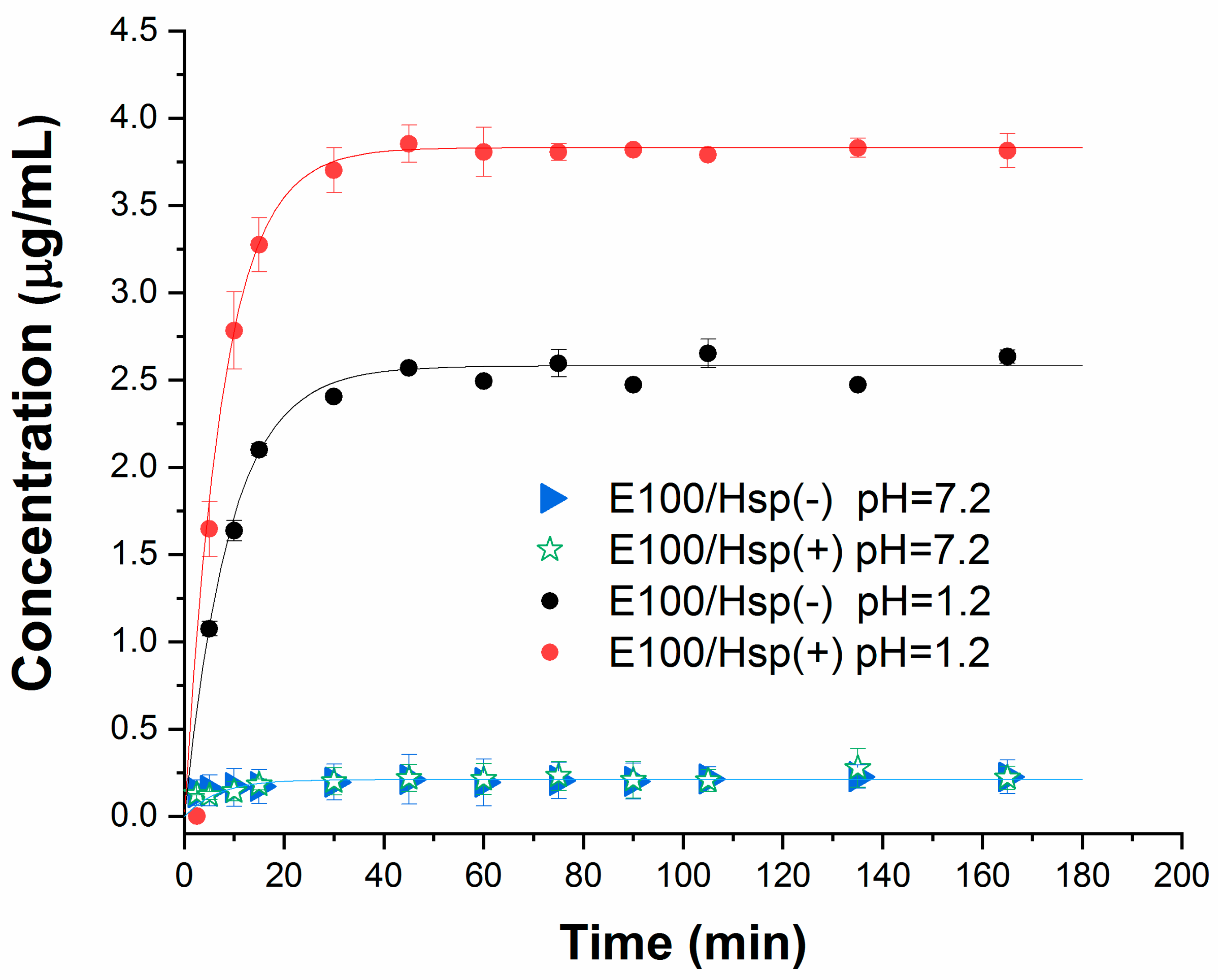
The results of the drug dissolution tests in simulated gastric and intestinal fluids (pH = 1.2 and pH = 7.2, respectively) are shown in
Figure 11. In each solution, the drug release profiles were verified from E100/Hesperidin filament samples at low (E100-Hsp(−)) and high (E100-Hsp(+)) concentration levels.
It is possible to verify that both samples with pharmaceutical form have a profile of rapid and complete release of Hsp in an acid medium; however, the polymeric matrix is shown as a device to protect the drug in a basic medium. This behavior is like that of the polymer in its original form, confirming that the extrusion process does not change the characteristics of the copolymer for usage as a filament in the release of active principles applications.
This rapid dissolution of the Hesperidin in acid medium, considering the diffusion of the drug along cylindrical matrices [
48], can be conveniently modeled by the Weibull function (continuous lines in the graph),
where
and
represent the drug concentrations in the medium at time t and the final time, respectively,
represents a time scale parameter, and
represents the dissolution kinetics degree. The drug release from the E100/Hsp(−) and E100/Hsp(+) matrices returned the value of
= 1, showing exponential kinetics, typical of delivery systems with a rapid drug-release response.
Thus, while the total release of the drug is reached in approximately 30 min in the simulated gastric fluid, for both concentrations tested, the release of the active principle in the intestinal fluid is minimized, even with a longer analysis time interval. These results prove the pH-dependent characteristic of E100 (drug protection in basic media and dissolution in acidic media) are retained when in the form of 3D printing filaments. It is important to highlight that the polymer release profile is not interfered with by protonation. Protonation interferes with the flexibility of the filament, based on the best molecular arrangement in the polymer chain, as discussed.
In addition, a pharmaceutical solid solution model part (10 mm in diameter and 3 mm in height) was designed in a CAD environment (
Figure 12a) and then printed (
Figure 12b) using the E100/Hsp(−) filament (arranged on a Kywoo3D Tycoon commercial direct drive 3D printer). The part was printed using Repetier Host slicer software v. 2.2.4, with a grid-type infill pattern (18 layers, 60% inner density), extrusion temperature of 130 °C, and printing time of 4.5 min.
The regular diameter and flexibility of the filament, in addition to the printing settings in the slicer software, were suitable for the layered material deposition process to occur, without clogging the printing system (due to possible filament breaks), and with physical reproduction of the designed part.
Figure 12c shows the circular section of the sample, in the sixteenth layer of molten filament deposition. In this layer, it is possible to observe the inner infill pattern, with the formation of parallel lines spaced apart across the entire section of the object. This configuration is repeated throughout the entire interior of the part, forming a grid-like structure, as projected in the slicer software.
At the edge of the section, it is also possible to observe a continuous portion of the area filled with molten material as a result of the beginning of the transition from 60% filling density of the internal sections to a total filling of the object area in the eighteenth layer (sample surface).
Figure 12d shows a detail of the characteristic dispersion of the drug in the printed part, with a regular particle size distribution below 2 um. This result indicates that the largest particulate aggregates of Hesperidin tend to decrease considerably in size upon interaction with the polymeric matrix, which favors the bioavailability of the active ingredient.

 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}