
Abstract
Anaplasmataceae agents are obligatory intracellular Gram-negative α-proteobacteria that are transmitted mostly by arthropod vectors. Although mammals of the Superorder Xenarthra (sloths, anteaters, and armadillos) have been implicated as reservoirs for several zoonotic agents, only few studies have sought to detect Anaplasmataceae agents in this group of mammals. This study aimed to investigate the occurrence and genetic diversity of Anaplasma spp. and Ehrlichia spp. in blood and spleen samples of free-living Xenarthra from four different states in Brazil (São Paulo, Mato Grosso do Sul, Rondônia, and Pará). Nested and conventional PCR screening assays were performed to detect the rrs and dsb genes of Anaplasma spp. and Ehrlichia spp., respectively. The assays were positive in 27.57% (91/330) of the Anaplasma spp. and 24.54% (81/330) of the Ehrlichia spp. Of the 91 positive Anaplasma spp. samples, 56.04% were positive in a conventional PCR assay targeting the 23S–5S intergenic region. Phylogenetic and distance analyses based on the rrs gene allocated Anaplasma sequences from sloths captured in Rondônia and Pará states in a single clade, which was closely related to the A. marginale, A. ovis, and A. capra clades. The sequences detected in southern anteaters from São Paulo were allocated in a clade closely related to sequences of Anaplasma spp. detected in Nasua nasua, Leopardus pardalis, and Cerdocyon thous in Brazil. These sequences were positioned close to A. odocoilei sequences. Genotype analysis corroborated previous findings and demonstrated the circulation of two distinct Anaplasma genotypes in animals from north and southeast Brazil. The first genotype was new. The second was previously detected in N. nasua in Mato Grosso do Sul state. The intergenic region analyses also demonstrated two distinct genotypes of Anaplasma. The sequences detected in Xenarthra from Pará and Rondônia states were closely related to those in A. marginale, A. ovis, and A. capra. Anaplasma spp. sequences detected in Xenarthra from São Paulo and were allocated close to those in A. phagocytophilum. The analyses based on the dsb gene grouped the Ehrlichia spp. sequences with sequences of E. canis (São Paulo, Mato Grosso do Sul, and Pará) and E. minasensis (Rondônia and Pará). The data indicate the occurrence of E. canis and E. minasensis and two possible new Candidatus species of Anaplasma spp. in free-living mammals of the Superorder Xenarthra in Brazil.
Similar content being viewed by others

Introduction
The Anaplasmataceae family (order Rickettsiales) is formed by the genera Anaplasma, Ehrlichia, Neorickettsia, and Wolbachia1. They are comprised of obligatory intracellular Gram-negative bacteria, that transmit mostly through arthropod vectors, mainly ticks. These agents can infect different mammalian cells depending on the infecting species. A. marginale, A. centrale, and A. ovis infect erythrocytes; A. platys infects platelets and neutrophils; A. odocoilei infects platelets; A. phagocytophilum and E. ewingii infect granulocytes; A. bovis, E. chafeensis, E. canis, E. muris, and Neorickettsia spp. infect monocytes and macrophages; E. ruminantium infects endothelial cells, neutrophils, and macrophages; and Wolbachia spp. is an invertebrate endosymbiont1,2. They form microcolonies (morulas) in intracytoplasmic vacuoles3. Domestic and wild animals are considered reservoirs, and favor the spread of these agents that can cause diseases in animals and humans1,3,4.
In Brazil, several genotypes of A. phagocytophilum, A. marginale, A. bovis, A. platys, E. canis, E. chafeensis, and potential new species, that are not yet named, have been detected in different species of wild animals, including deer5,6,7,8,9,10,11, wild canids12,13,14, wild felids12,15,16, coatis14, rodents14,17,18,19, caititus and peccaries20, opossums21,22, and birds23,24.
The Xenarthra superorder is composed of two orders: Cingulata and Pilosa. Cingulata is characterized by the presence of a bone carapace. A representative animal is the armadillo. Pilosa features a dense coat and the absence or underdevelopment of teeth. Representative animals are sloths and anteaters25,26. These animals are distributed from the central southern region of North America to southern of South America26. They are considered reservoirs of several zoonotic agents27.
Studies on Anaplasmataceae agents in Xenarthra are scarce. Anaplasma marginale was detected in a giant anteater (Myrmecophaga tridactyla) in Argentina by blood smears and molecular confirmation by PCR assays based on the msp-5 and msp1-α genes28. Genotypes of Ehrlichia spp., based on the dsb gene, and Anaplasma spp., based on the 16S rRNA gene, were detected in a three-toed sloth (Bradypus tridactylus) in the state of Pará in northern Brazil30.
The present study aimed to investigate the occurrence and genetic diversity of Anaplasma spp. and Ehrlichia spp. in Xenarthra mammals sampled in four different Brazilian states.
Material and methods
Ethical statement
Animal procedures and management protocols were approved by the Institute Chico Mendes for Conservation of Biodiversity (SISBIO N º 53798-5) and by the Ethics Committee on Animal Use of the Biomedical Sciences Institute—University of São Paulo (protocol number 98) and the School of Agricultural and Veterinarian Sciences (FCAV/UNESP; protocol number 11.794). All methods and experimental protocols were performed in accordance with the relevant FCAV/UNESP Ethics Committee guidelines and regulations.
Sampling and sampled species
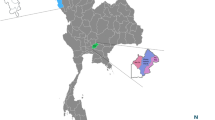
The biological samples of Xenarthra mammals originated from four Brazilian states: Mato Grosso do Sul (MS), São Paulo (SP), Pará (PA), and Rondônia (RO). The total of 330 animals included 188 brown-throated sloths (Bradypus variegatus), three Bradypus spp., five two-toed sloths (Choloepus didactylus), 31 Choloepus spp., 31 southern anteaters (Tamandua tetradactyla), 52 Giant Anteaters (Myrmecophaga tridactyla), three southern naked-tailed armadillos (Cabassous unicinctus), eight nine-banded armadillos (Dasypus novemcinctus), eight six-banded armadillo (Euphractus sexcinctus), and one giant armadillo (Priodontes maximus) (Fig. 1).

Number and origin of the mammals from the Superorder Xenarthra sampled in Brazil. SP São Paulo, MS Mato Grosso do Sul, PA Pará, RO Rondonia. The map was created using QGIS v.3.10.5 software (https://www.qgis.org/en/site/forusers/download.html.).
Spleen samples were collected by the Bandeiras e Rodovias Project team and by the Wildlife Pathology Service of FCAV/UNESP during necropsies of road-killed animals in the Cerrado biome in Mato Grosso do Sul (20 °26′ 48.3″ S 52° 54′ 11.6″ W) and São Paulo (21° 17′ 33.2″ S 48° 19′ 53.4″ W). In the state of Mato Grosso do Sul, 55 spleen samples (33 giant anteaters, eight southern anteaters, eight six-banded armadillos, three southern nine-banded armadillos, one giant armadillo, and two southern naked-tailed armadillos) were collected during 2017 and 2018. In São Paulo state, 39 spleen samples (19 giant anteaters, 16 southern anteaters, and four southern nine-banded armadillos) were obtained from 2011 to 2018. In addition, 236 blood samples were collected by cephalic vein venipuncture from sloths, armadillo and anteaters during the filling process of a hydroelectric power plant in Amazonia biome in the states of Rondônia (3° 07′ 20.2″ S 51° 46′ 31.5″ W) (n = 102, comprising 65 brown-throated sloths, 31 Choloepus spp., three Bradypus spp., one giant anteater, one nine-banded armadillo, and one southern naked-tailed armadillo) and Pará (9° 16′ 21.1″ S 64° 37′ 59.2″ W) (n = 134, comprising 123 brown-throated sloths, five two-toed sloths, and six giant anteaters).
Spleen fragments and blood samples were stored in KASVI® RNAse and DNAse-free microtubes at − 70 °C until DNA extraction.
Molecular analysis
DNA was extracted from 10 mg of each spleen tissue and 200 μL of each blood sample using the DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA, USA), according to the manufacturer’ s instructions. The presence of amplifiable DNA was verified by a conventional PCR (cPCR) assay targeting the mammalian endogenous glyceraldehyde-3-phosphate dehydrogenase (gapdh) gene29. The positive samples were subjected to specific PCR assays for Anaplasma spp. and Ehrlichia spp. (Fig. 2).

Workflow with the steps performed in the methodology of this study.
Anaplasmaspp.detection
Screening for Anaplasma spp. based on the groEL and rrs genes was performed using multiplex qPCR (quantitative real-time PCR)18 and nested PCR (nPCR)30 protocols, respectively (Tables 1, 2). The positive samples, for at least one of the two protocols, were tested by qPCR assays based on the msp-231 and msp1-β32 genes for A. phagocytophilum and A. marginale, respectively (Table 1). cPCR assays for Anaplasma spp. based on the 23S–5S intergenic region were also performed33 (Table 2).
Ehrlichiaspp.detection
Screening for Ehrlichia spp. based on groEL and dsb genes was performed using multiplex qPCR (quantitative real-time PCR)18 and cPCR34 protocols, respectively (Tables 1, 2). The positive samples, for at least one of the two protocols, were tested by qPCR assays based on dsb34 and vlpt35 genes for E. canis and E. chaffeensis, respectively (Table 1). Additionally, cPCR assays for Ehrlichia spp. were based on the groEL36 and sodB genes37. In addition, nPCR assays for Ehrlichia spp. based on the omp-1 gene38, were performed (Table 2).
Reactionconditions
All qPCR assays were performed with a final volume of 10 μL containing 1 μL of DNA sample (concentration mean: 162.5 and 50.55 ng/µL for spleen and blood samples, respectively), 0.2 μM of each primer and hydrolysis probe, 5 μL GoTaq Probe qPCR Master Mix (Promega Corporation, Madison WI, USA), and sterilized ultrapure water (Nuclease-Free Water; Promega Corporation) q.s. 9 μL. PCR amplifications were performed in low-profile multiplate unskirted PCR plates (Bio-Rad, Hercules, CA USA) using a CFX96 Thermal Cycler (Bio-Rad). Quantification of the number of copies of target DNA/μL was performed using IDT psmart plasmids (Integrated DNA Technologies, Coralville, IA, USA) containing the target sequences. Serial dilutions were performed to construct standard curves with different plasmid DNA concentrations (2.0 × 107 to 2.0 × 100 copies/μL). The number of plasmid copies/µL of the amount (g/µL) of DNA/plasmid (bp) was determined by multiplying by 6.022 × 1023. Each qPCR assay was performed in duplicate for each DNA sample. All duplicates showing cycle quantification (Cq) values differing by > 0.5 were re-tested. Amplification efficiency (E) was calculated from the slope of the standard curve in each run (E = 10–1/slope). The reactions followed the standards established by the Minimum Information for Publication of Quantitative real-time PCR experiments39.
All the cPCR assays were performed using 5 μL of the DNA samples (concentration mean: 162.5 and 50.55 ng/µL for spleen and blood samples, respectively) in a mixture containing 1.25 U Platinum Taq DNA Polymerase (Invitrogen, Carlsbad, California, USA), PCR buffer (PCR buffer 10×—100 nM Tris–HCl, pH 9.0, 500 mM KCl), 0.2 mM deoxynucleotides (dATP, dTTP, dCTP, and dGTP) (Invitrogen, Carlsbad, California, United States), 1.5 mM of magnesium chloride (Invitrogen, Carlsbad, CA, United States), 0.5 μM of each primer (Invitrogen), and sterile ultrapure water (Invitrogen) q.s.25 μL. In nPCR assays, 1 μL of the amplified product from the first PCR reaction was used as the target DNA in the second reaction. DNA samples from A. phagocytophilum, kindly provided by Prof. John Stephen Dumler (Uniformed Services University of the Health Sciences, Bethesda, MD, USA), and E. canis, obtained from DH82 cells infected with the Jaboticabal strain of E. canis40, were used as positive controls. Sterile ultrapure water (Nuclease-Free Water, Promega Corporation) was used as a negative control. The products were separated by 1% agarose gel electrophoresis at an electric current of 100 V/150 mA for 50 min. The gel was stained with 1% ethidium bromide (Life Technologies, Carlsbad, CA, USA) and examined under ultraviolet light illumination using the ChemiDoc MP Imaging System (Bio-Rad) and photographed using Image Lab Software version 4.1.
The cPCR and nPCR amplified products were purified using the ExoSAP-IT PCR Product Cleanup Reagent (Applied Biosystems, Foster City, CA, USA) and sequenced using the BigDye Terminator v3.1 Cycle Sequencing kit (Thermo Fisher Scientific, Waltham, MA, USA) and the ABI PRISM 310 DNA Analyzer (Applied Biosystems)41.
Phylogenetic analyses
The sequences obtained were submitted to a quality-screening test using Phred-Phrap software (version 23)42,43 to evaluate the quality of the electropherograms and to obtain the consensus sequences from the alignment of the sense and antisense sequences. The BLASTn program44 was used to compare the obtained nucleotide sequences with previously deposited sequences in the GenBank database45.
The consensus sequences obtained in this study and those retrieved from GenBank were aligned using ClustalW software version 746 using Bioedit v. 7.0.5.347. The best evolutionary model was chosen using the jModelTest2 software (version 2.1.6) on XSEDE48 via the CIPRES Science Gateway49. The phylogenetic analyses were based on Bayesian inference (BI) and maximum likelihood (ML) methods. The BI analyses were performed using MrBayes 3.1.250 via the CIPRES Science Gateway. Markov chain Monte Carlo simulations were run for 106 generations with a sampling frequency of every 100 generations and a 25% burn-in. ML analyses were performed using the Blackbox RaxML cluster51 using 1,000 bootstrapping replicates52. The phylogenetic trees were edited using TreeGraph 2.0.56-381 beta software53.
Genetic diversity and genealogies
The genetic diversity analyses for the rrs gene and 23S–5S intergenic region of Anaplasma spp. and for the dsb gene of Ehrlichia spp. were performed with the sequences obtained in this study aligned to phylogenetically closer sequences of A. phagocytophilum, A. marginale, A. ovis, A. odocoilei, Anaplasma spp., E. canis, E. minasensis, and Ehrlichia spp. retrieved from GenBank. Clustal/W software46 via Bioedit v. 7.0.5.347 was used for the alignment. The sequences used were at least 420 bp, 310 bp, and 310 bp for the rrs gene, 23S–5S intergenic region, and dsb gene, respectively. Sequences that were smaller in size were excluded from the phylogenetic analysis. These alignments were used to calculate the nucleotide diversity (π), polymorphism level (diversity of haplotypes [Dh], number of haplotypes [h], and the average number of nucleotide differences [K]), using DnaSP v5 software54. The sequences were submitted to the TCS Network55 and distance analysis based on the split-network was inferred using the programs Population Analysis with Reticulate Trees (popART)56 and Splitstree v 4.14.6 57, respectively.
Results
Occurrence of Anaplasmataceae agents in Xenarthra mammals
The mean concentration of the extracted DNA was 162.5 ± 37.8 and 50.55 ± 12.1 ng/µL for spleen and blood samples, respectively. All 330 DNA samples were positive in the cPCR assay based on the endogenous gapdh gene. A total of 147 (44.54%) animals were positive for at least one agent.
Financial restraints limited the selection of samples for sequencing to only a few among the large number of positive samples. The selection was based on two steps. First, we selected samples that presented high intensity amplicons in agarose gel electrophoresis. These samples were then separated according to animal species and region of origin. A random selection was performed to obtain at least one representative of each positive species and of each location.
Anaplasma spp.
Of the 330 samples analyzed, 91 (27.57%) were positive for Anaplasma spp. based on the rrs gene (Table 3). No sample was positive in the qPCR for Anaplasma spp. based on the groEL gene. Of the 91 samples positive in the screening assays for Anaplasma spp., 51 (56.04%) samples were positive for the 23S–5S intergenic region of Anaplasma spp., and 7/91 (7.7%) samples were positive in the qPCR assay for A. phagocytophilum based on the msp-2 gene. The latter positive samples were not quantified because of the low amount of DNA of the agent in the tested samples (Monte Carlo effect)42 (Table 3). All samples were negative in the qPCR assays for A. marginale (msp1-β gene) (Supplementary Table 1).
rrsgene
Of the 91 samples, 25 (27.47%) positive samples for Anaplasma spp., based on the rrs gene, were sequenced. The sequences obtained were deposited in GenBank under accession numbers MT199810 to MT199833.
BLASTn analysis showed that nine rrs Anaplasma sequences obtained from animals sampled in Rondônia and Pará states showed identity ranging from 97.39 to 98.48% with sequences of A. phagocytophilum detected in Haemaphysalis longicornis (GU064895) and in Chinese water deer (Hydropotes inermis; KR611598) sampled in Korea. The 16 remaining sequences of animals from the same states showed identities ranging from 97.71 to 99.38% with Anaplasma spp. genotypes detected in Rattus rattus from Brazil (KY391803) and in a goat from Saudi Arabia (LC467273). Two sequences detected in Xenarthra sampled from São Paulo state showed identities of 99.04 and 99.50% with Anaplasma spp. detected in a Brazilian Brown Brocket Deer (Mazama gouazoubira; KF020580) and in a coati (N. nasua; KY499186) from Brazil, respectively. One sequence from São Paulo showed 98.69% identity with A. phagocytophilum detected in a dromedary from Tunisia (KC455363) (Table 4).
The phylogenetic analysis inferred by the BI method (Fig. 3a) positioned all the sequences obtained in Rondônia and Pará states in a single clade that was phylogenetically closer to Anaplasma spp. genotypes detected in rodents in Brazil (KP757841 and KY391803), with 89% branch support. Despite forming a single clade, the obtained sequences showed greater proximity to the clade of Anaplasma spp. found in ruminants (A. capra, A. ovis, and A. marginale). On the other hand, the sequences obtained from anteaters of São Paulo state were allocated to a clade closer to the sequences of Anaplasma spp. detected in ocelots (Leopardus pardalis), coatis (Nasua nasua), and crab-eating foxes (Cerdocyon thous) from the Pantanal natural region in southern Brazil, with 87% branch support. This clade was sister to the clade of A. odocoilei, with 90% branch support. Results of the ML analysis (Fig. 3b) concurred (or agreed) partially with the BI analysis. While the sequences obtained in Rondônia and Pará states formed a single clade close to the clade of Anaplasma spp. found in ruminants, the Anaplasma sequences obtained from Xenarthra of São Paulo were subdivided into different clades, allocated close to the clade of A. odocoilei.

Phylogenetic analysis of Anaplasma rrs sequences based on the topology generated by Bayesian model (A) and Maximum Likelihood (B), with TVM + I + G as evolutionary model. Ochrobactrum anthropi, Brucella melitensis and Mesorhzobium Ioti were used as an external group.
Additionally, four (BM31, BM32, BM128, and BM129) of the seven positive samples for A. phagocytophilum, based on the msp2 gene, were sequenced for the rrs gene. Interestingly, three sequences had a high identity with A. phagocytophilum by the BLASTn analysis. However, in the phylogenetic analysis, they were positioned together with rrs sequences of other Anaplasma spp. obtained in Rondônia and Pará states. BLASTn directly presented a greater identity of one sequence (BM31) with Anaplasma sp. of Rattus rattus.
Genotype analysis based on 38 rrs Anaplasma sequences, including 18 sequences obtained in this study and sequences of A. marginale, A. ovis, A. phagocytophilum, A. odocoilei, and Anaplasma spp., indicated the presence of 12 genotypes, with π = 0.01719, hd = 0.808, and K = 7.16643 (Table 5, Fig. 4a). Eight genotypes comprised more than one sequence. Genotype #3 comprised two sequences of A. phagocytophilum detected in South Korea. Genotype #4 comprised 16 sequences detected in sloths from Rondônia and Pará (this study). Genotype #5 comprised three sequences of A. phagocytophilum detected in the United States and Norway. Genotype #6 grouped two sequences of A. marginale detected in cattle from the Philippines and Uganda. Genotype #7 grouped two sequences of A. ovis detected in sheep and deer in China. Genotype #8 grouped the two sequences detected in anteaters from São Paulo state (this study) and one sequence detected in a coati (Nasua nasua) from MS. Genotype #11 comprised three sequences of A. odocoilei sequences detected in cervids and a fly (Lipoptena depressa) in the United States. Finally, genotype #12 grouped sequences of Anaplasma sp. detected in ocelot (Leopardus pardalis), crab-eating fox (Cerdocyon thous), and coati from Brazil.

(A) TCS network of rrs sequences from A. marginale, A. phagocytophilum, A. ovis and those obtained in the present study performed with PopART v.1.7 software (https://popart.otago.ac.nz/index.shtml) (B) Split-network performed with Splitstree v4.14.6 software using the parameters “Neighbor-Net and” Uncorrected p-distance (https://en.freedownloadmanager.org/Windows-PC/SplitsTree4-FREE.html.).
Regarding the genotype network (Fig. 4a), it could be inferred that genotype #4, which comprises the rrs sequences of Anaplasma detected in sloths from Rondônia and Pará states, was derived from genotype #1, which comprises a sequence found in Rattus in Brazil (KY391803), upon a mutational event. Both genotypes originated from a median vector (genotype not contemplated in the presented tree). On the other hand, genotype #8, which covers the two sequences obtained in this study in anteaters from São Paulo state and a coati sequence obtained in the state of MS, originated from a median vector upon a mutational event. Moreover, the sequences of A. odocoilei also originated from the same median vector. Additionally, genotype #8 gave rise to genotype #12, composed of sequences obtained from different wild animals sampled in the state of MS.
Split-network analysis based on the rrs gene (Fig. 4b) corroborated the genotype network, since the Rondônia and Pará sequences were all positioned together and closer to the Anaplasma spp. sequence previously detected in R. rattus. The phylogenetic analysis revealed that they were positioned closer to the A. ovis sequences. The sequences detected in São Paulo were closely positioned to the sequences of A. odocoilei.
23S–5Sregionintergenic
Fourteen (27.45%) of 51 positive samples for Anaplasma spp. based on the 23S–5S intergenic region were sequenced. The sequences obtained were deposited in GenBank under access numbers MT267341 to MT267354.
BLASTn analysis showed that 12 sequences obtained in Rondônia and Pará states showed identity ranging from 90 to 90.91% with A. marginale detected in a bovine animal from Brazil (CP023731), and two sequences from São Paulo showed 90.58 and 90.74% identity with A. phagocytophilum detected in a sheep from Norway (CP015376) (Table 4).
Phylogenetic analyses based on the 23S–5S intergenic region of Anaplasma spp. positioned the sequences detected in Xenarthra in two distinct clades, composed only of sequences found in this study. The first clade was composed of the sequences detected in sloths from the Rondônia and Pará states, which was in close proximity to the clade of A. marginale and A. ovis. The second clade was composed of sequences obtained from anteaters from SP, which were in close proximity to the clade of A. phagocytophilum. Both the BI (Fig. 5a) and ML analyses (Fig. 5b) presented the same topology, although ML presented a better definition of the clades. The index of clade support was 100 and 99% for Rondônia and Pará clade 1 and 89 and 100% for SP clade 2, in the BI and ML analyses, respectively. Of the seven (7.7%) positive samples in the qPCR for the msp2 gene of A. phagocytophilum, only one (14.28%) was positive in the PCR based on the 23S–5S region. However, it was closer to A. marginale in both the BLASTn and phylogenetic analyses.

Phylogenetic analysis of Anaplasma 23S–5S sequences based on the topology generated by Bayesian (A) and Maximum Likelihood (B) models, with HKY + G as evolutionary model. Ehrlichia muris and Ehrlichia chaffeensis were used as an external group.
Genotype analysis based on 23 Anaplasma 23S–5S sequences, which included 14 sequences obtained from the Xenarthra sampled in this study as well as A. marginale, A. ovis, and A. phagocytophilum sequences, obtained eight different genotypes, with π = 0.08454, hd = 0.719, and K = 24.09486 (Table 5, Fig. 6a). Four genotypes comprised more than one sequence. Genotype #1 comprised all 12 Anaplasma 23S–5S sequences from sloths sampled in Rondônia and Pará states. Genotype #8 comprised two sequences detected in giant anteaters from São Paulo state. Genotype #2 comprised three sequences of A. marginale previously detected in Brazil and Mexico. Genotype #5 comprised two sequences of A. phagocytophilum detected in the United States and Norway. Based on the network of genotypes (Fig. 6a), genotype #1 originated from a median vector and several mutational events. The same median vector also originated from genotypes #2 and #4, corresponding to sequences of A. marginale. Genotype #8 also originated from a median vector, which in turn originated from genotype #7, and gave rise to genotypes #5 and 6, comprised of A. phagocytophilum sequences.

(A) TCS network of 23S–5S sequences from A. marginale, A. phagocytophilum, A. ovis and those obtained in the present study performed with PopART v.1.7 software (https://popart.otago.ac.nz/index.shtml) (B) Split-network performed with Splitstree v4.14.6 software using the parameters “Neighbor-Net and” Uncorrected p-distance (https://en.freedownloadmanager.org/Windows-PC/SplitsTree4-FREE.html.).
Corroborating the genotype network findings, the split-network analysis, based on the 23S–5S intergenic region (Fig. 6b), showed a clear separation between the sequences detected in Rondônia and Pará states, and those detected in São Paulo state. Additionally, the Xenarthra sequences from Rondônia and Pará states were closer to the A. marginale and A. ovis sequences. The Xenarthra sequences from São Paulo were allocated close to A. phagocytophilum, corroborating the findings obtained in the other analyses.
Ehrlichia spp.
Of the 330 samples screened for Ehrlichia spp., 81 (24.54%) were positive in cPCR assays based on the dsb gene (Table 3). No sample was positive in qPCR for Ehrlichia spp. based on the groEL gene. No samples were positive in the cPCR (groEL and sodB genes), in the nested PCR (omp-1 gene) assays for Ehrlichia spp., and in the qPCR assays for E. canis (dsb gene) and E. chaffeensis (vlpt gene) (Supplementary Table 2).
Nineteen (23.45%) of 81 samples positive for Ehrlichia spp. PCR based on the dsb gene were sequenced. The sequences obtained were deposited in GenBank under access numbers MT212405 to MT212423.
BLASTn analysis showed that 10 sequences were identical to E. canis detected in a dog from Colombia (MK783026). The remaining nine sequences showed identities ranging from 98.96 to 100% with E. minasensis detected in Rhipicephalus microplus from Brazil (JX629808) and in bovine from Australia (MH500007) (Table 4).
Phylogenetic analyses based on the dsb gene of Ehrlichia spp. positioned the sequences obtained in this study in the E. canis and E. minasensis clades. The sequences obtained from São Paulo and Mato Grosso do Sul, and three from Pará were grouped in the clade of E. canis, and four sequences from Pará and all from Rondônia were grouped in the clade of E. minasensis. The topologies of phylogenetic trees obtained by both BI (Fig. 7a) and ML (Fig. 7b) methods corroborated the findings. Additionally, ML analysis inferred a subdivision within the E. canis clade. Five sequences found in Xenarthra mammals formed a minor clade close to a sequence detected in a dog from Colombia. Four sequences formed a second clade with the other sequences of E. canis analyzed. Finally, a sequence obtained from a M. tridactyla (83) from São Paulo was positioned separately from the others, with 98% branch support.

Phylogenetic analysis of Ehrlichia dsb sequences based on the topology generated by Bayesian model (A) and Maximum Likelihood (B), with TrN + I + G as evolutionary model. Ehrlichia muris and Ehrlichia chaffeensis were used as an external group.
Genotypes based on 29 Ehrlichia sequences were analyzed. These included 17 sequences obtained in this study as well as E. canis and E. minasensis sequences detected in different countries. A total of four genotypes were found, with π = 0.03570, hd = 0.569, and K = 9.28079 (Table 5). Genotype #1 comprised all sequences of E. canis retrieved from GenBank as well as the sequences detected in Xenarthra sampled in this study in São Paulo and Mato Grosso do Sul, and three sequences from Pará (BM24, BM51, and BM61). Genotype #2 comprised all E. minasensis sequences retrieved from GenBank, three Xenarthra sequences sampled in Rondônia, and four from Pará (BM16, BM177, BM180, PV14). Genotypes #3 and #4, on the other hand, comprised unique sequences detected in specimens of B. variegatus from Rondônia, PV337, and PV41, respectively. Based on genotype network analysis (Fig. 8a), genotype #1 (E. canis) originated from genotype #3 through several mutational events. The latter seems to have originated from genotype #2 (E. minasensis), which, in turn, originated from genotype #4, both from a mutational event. The split-network analysis corroborated the main findings described by the genotype network analysis (Fig. 8b).

(A) TCS network of dsb sequences of E. canis, E. minasensis and those obtained in the present study performed with PopART v.1.7 software (https://popart.otago.ac.nz/index.shtml (B) Split-network performed with Splitstree v4.14.6 software using the parameters “Neighbor-Net and” Uncorrected p-distance (https://en.freedownloadmanager.org/Windows-PC/SplitsTree4-FREE.html.).
Co-positivity for Anaplasma spp. and Ehrlichia spp.
Of the 147 positive animals, 25 (17%) were co-positive for Anaplasma spp. and Ehrlichia spp., among 21 sloths (18 B. variegatus [12 from Pará and six from Rondônia], one C. didactylus from Pará, one Bradypus spp. and one Choloepus spp. from Rondônia), and four anteaters (three T. tetradactyla and one M. tridactyla from São Paulo).
Discussion
The present study revealed a high rate of positivity for Anaplasma spp. and Ehrlichia spp. in Xenarthra mammals sampled in four different Brazilian states. Of the 330 animals, 147 (44.54%) were positive for at least one of the agents. Of these, 25 (17%) were positive for both Ehrlichia spp. and Anaplasma spp. Until this study, molecular data concerning Anaplasmataceae agents in biological samples of the Superorder Xenarthra have been scant. Guillemi et al.28 detected the presence of A. marginale morulae in blood smears from a giant anteater in Argentina, which was confirmed by PCR assays based on msp-5 and msp1-α genes. In Brazil, Soares et al.20 detected a new dsb genotype of Ehrlichia sp. in a three-toed sloth (Bradypus tridactylus) obtained in the state of Pará. The sequence was allocated a separate clade that was sister to the clade of E. ruminantium, and close to the sequences of Ehrlichia spp. detected in a horse and a fox in Brazil. The same animal was positive for the rrs gene, whose sequence was allocated in a clade close to A. phagocytophilum.
The present study reports the occurrence of two possible species of Anaplasma spp. in mammals of the Superorder Xenarthra from Brazil, since the two genes analyzed showed low identity values obtained by BLASTn and the phylogenetic findings positioned the sequences obtained in this study in single clades that were separate from the others.
BLAST analyses performed for Anaplasma spp. showed that all rrs sequences detected in sloths from the states of Rondônia and Pará showed identity values lower < 99% (not exceeding 98.5%) with sequences of A. ovis, A. marginale, and A. centrale. Additionally, the sequences detected in anteaters from São Paulo also showed an identity < 99% with sequences of A. phagocytophilum and A. odocoilei. However, these same sequences showed an identity > 99% with an Anaplasma sequence previously detected in a coati from MS (KY4999186). Previous studies have defined rrs sequences as having at least 95% identity to be identified at the genus level and 99% to be identified at the species level58,59,60. In view of this, we have proposed two new species circulating in these animals, and the species detected in São Paulo’s anteaters is probably the same as that found in the Mato Grosso do Sul coatis. The analyses performed in the 23S–5S region intergenic sequences corroborated with the rrs gene. The identities obtained were quite low, not exceeding 90%, strengthening the hypothesis of two new species.
The phylogenetic analyses corroborated the BLASTn results. The analysis of the rrs sequences of Anaplasma spp. obtained from sloths of Rondônia and Pará were allocated close to two Anaplasma sequences previously detected in rodents (R. rattus and H. megacephalus) from Brazil. In addition, the clades formed by these two sequences and those found in the present study had a sister clade formed by A. marginale, A. ovis, and A. capra. The Anaplasma sequences detected in anteaters from São Paulo were allocated in a clade close to a new genotype of Anaplasma spp. previously detected in wild mammals from the Pantanal Sul-matogrossense.
A similar topology was observed in the phylogeny based on the 23S–5S intergenic region, in which the clade formed by the Anaplasma sequences from Xenarthra sampled in Rondônia and Pará was a sister clade that was formed by Anaplasma species detected in ruminants. The two Anaplasma sequences obtained from anteaters in the state of São Paulo were in a clade completely separated from the other sequences, although they were closer to the A. phagocytophilum clade, raising questions about the possible influence of the geographical location or host species on the occurrence of Anaplasma species that affect these animals.
Out of the seven samples positive in the qPCR for the msp2 gene of A. phagocytophilum, only one (14.2%) was positive in the PCR based on the 23S–5S region. Despite this, it was phylogenetically related to the clade formed by A. marginale and A. ovis. Similarly, four samples that were also positive for the msp2 gene were positioned close to the clade of A. marginale, A. ovis, and A. capra in the phylogenetic analysis based on the rrs gene. This potentially indicates the possibility of the aforementioned qPCR protocol to amplify msp-2 gene fragments from Anaplasma species phylogenetically related to A. phagocytophilum. Alternatively, the animals may have been co-infected with A. phagocytophilum and the new Candidatus species. MSP2, an external membrane protein present in all Anaplasma species, is encoded by several polymorphic genes in A. marginale, A. centrale, and A. ovis, and by only one gene in A. phagocytophilum61.
To better understand the results, genetic diversity analysis and distance genealogies were performed. The results of both corroborated the previously presented phylogenetic positioning. For both the rrs gene and the 23S–5S intergenic region, the genotype analyses showed that the Anaplasma sequences obtained in Rondônia and Pará states formed new genotypes (#4 and #1, for rrs and intergenic regions, respectively), whereas the sequences obtained from anteaters in the SP state comprised one genotype (#8 for rrs and intergenic region).
In addition, the genotype network based on the rrs gene suggests that the genotype circulating in sloths in Rondônia and Pará states might have originated through three mutational events from genotype #1, which was detected in a R. rattus from Brazil, explaining the proximity of both in phylogenetic analysis. The genotype circulating in São Paulo anteaters is the same genotype previously found in a coati in Mato Grosso do Sul, which might have originated through a mutational event from a median vector. Additionally, the genotype network based on the intergenic region suggests that both genotypes (#1 and #8) found in this study might have originated from different median vectors through numerous mutational events. The distance analysis for both genes showed that Anaplasma sequences obtained from Xenarthra sampled in the northern region of the country were positioned apart from the others, but were closer to A. marginale and A. ovis. On the other hand, the sequences obtained from Xenarthra sampled in São Paulo were positioned apart, albeit closer to A. odocoilei and A. phagocytophilum, based on the rrs gene and intergenic region, respectively.
All analyses corroborated and provided strong evidence of the circulation of two new species of Anaplasma in Xenarhtra in Brazil, related to the region inhabited by these animals. We propose naming the species circulating in Rondônia and Pará states as ‘Candidatus Anaplasma amazonensis’ and the species detected in São Paulo as ‘Candidatus Anaplasma brasiliensis’. Further studies are needed to validate these species as well as to determine the vectors responsible for their transmission.
Regarding the findings for Ehrlichia spp., all analyses including BLASTn, corroborated and grouped the sequences as E. canis or E. minasensis. In addition, two sequences detected in sloths phylogenetically close to E. minasensis formed two new and distinct genotypes (#3 and #4).
The rrs genotypes phylogenetically related to E. canis have been increasingly described in wild animals from Brazil, and have already been detected in wild canids12,14, wild felids15, rodents14,18,19, coatis14, and geese (Neochen jubata)24. E. minasensis, a recently described species that is genetically similar to E. canis62,63, can infect and cause clinical signs in cattle in central-western Brazil64. It has already been detected in cattle in North America65, Ethiopia66, Brazil64, in cervids in Canada67, dogs in Israel68, and in several species of ticks68,69,70,71,72,73,74.
Interestingly, the dsb sequences of E. canis obtained from Xenarthra mammals comprised animals sampled in the states of São Paulo, Mato Grosso do Sul, and portions of Pará. The dsb sequences of E. minasensis were obtained from animals from Rondônia and Pará states. Similar to the analysis of rrs and 23S–5S Anaplasma sequences, these findings again raise questions about the possible regionalization of the Ehrlichia and Anaplasma species found in Xenarthra from Brazil.
Tick vectors of E. canis include R. sanguineus sensu lato (tropical lineage) and D. variabilis75,76,77. Although DNA from E. minasensis has already been detected in tick species that include Rhipicephalus (R. microplus and R. sanguineus)68,69,72,73, Hyalomma spp.71,72, Haemaphysalis hystricis74, and Amblyomma sculptum72, the vectorial competence and capability have not been assessed thus far.
The clear dichotomy between the Ehrlichia and Anaplasma species that infect Xenarthra from different regions of the country may be related to the distribution and abundance of tick species that function as Anaplasmataceae vectors78. However, previous studies performed in different Brazilian states have shown that the different tick species that parasitize Xenarthra mammals are more correlated to the host species than to the geographic region. For instance, sloths are mainly parasitized by Amblyomma varium and A. geayi, with the latter found more in the state of Pará. While giant anteaters and southern anteaters are usually parasitized by A. nodosum, A. sculptum, and A. calcaratum, armadillos are frequently parasitized by A. pseudoconcolor, A. auricularium, and A. sculptum27, 79,80,81,82. These studies show the high parasitic specificity of some species of ticks that infest these animals. For instance, the ticks collected from anteaters from the state of São Paulo in the present study were all identified as A. nodosum, which was the most frequently found species in this group of animals (data not shown).
The vectorial capacity of the tick species frequently found in Xenarthra mammals for Anaplasmataceae agents is still unknown. Although the majority of ticks that parasitize this group of mammals belong to the genus Amblyomma spp., the main vectors of Ehrlichia spp. in Brazil belong to the genus Rhipicephalus spp. Further studies should be conducted to assess the vectorial capacity of these ticks and to better understand the genetic diversity of Anaplasmataceae agents that infect mammals of the Xenarhra superorder in Latin America as well as the possible role of these animals in the epidemiological cycle of these agents.
Conclusion
The present study showed the high occurrence of Anaplasma spp. (27.57%) and Ehrlichia spp. (24.54%) in free-living Xenarthra mammals sampled from four states in Brazil. In addition, the study provides the first description of the occurrence of E. canis and E. minasensis in this group of mammals. The analysis of two genetic regions of Anaplasma spp., one conserved (rrs), and another one more diverse (intergenic region 23S–5S) revealed similar results of the low identity in BLASTn analysis, phylogenetic positioning in two different clades that were separate from the others containing known species, and formation of two different genotypes from those comprising known Anaplasma species by the diversity analysis. Based on these findings, we propose two new Candidatus species—‘Candidatus Anaplasma amazonensis’ and ‘Candidatus Anaplasma brasiliensis’—in Xenarthra from Brazil.
Data availability
The data tha support the findings of this study are openly available in National Center for Biotechnology Information at https://www.ncbi.nlm.nih.gov/, reference number rrs: MT199810–MT199833; dsb: MT212405–MT212423; 23S–4S intergenic region: MT267341–MT267354.
References
Dumler, J. S. et al. Reorganization of genera in the families Rickettsiaceae and Anaplasmataceae in the order Rickettsiales: unification of some species of Ehrlichia with Anaplasma, Cowdria with Ehrlichia and Ehrlichia with Neorickettsia, descriptions of six news species combinations and designation of Ehrlichia equi and “HGE agent” as subjective synonyms of Ehrlichia phagocytophila. Int. J. Syst. Evol. Microbiol. 51, 2145–2165 (2001).
Rar, V. & Golovljova, I. Anaplasma, Ehrlichia, and “Candidatus Neoehrlichia” bacteria: pathogenicity, biodiversity, and molecular genetic characteristics, a review. Infect. Genet. Evol. 11, 842–1861 (2011).
Doudier, B., Olano, J., Parola, P. & Brouqui, P. Factors contributing to emergence of Ehrlichia and Anaplasma spp. as human pathogens. Vet. Parasitol. 167, 149–154 (2010).
Parola, P., Davoust, B. & Raoult, D. Tick and flea-born rickettsial emerging zoonoses. Vet. Res. 36, 469–492 (2005).
Machado, R. Z., Duarte, J. M. B., Dagnone, A. S. & Szabó, M. P. J. Detection of Ehrlichia chaffeensis in Brazilian marsh deer (Blastocerus dichotomus). Vet. Parasitol. 139, 262–266 (2006).
Picoloto, G. et al. Real time polymerase chain reaction to diagnose Anaplasma marginale in cattle and deer (Ozotoceros bezoarticus leucogaster) of the Brazilian Pantanal. Rev. Bras. Parasitol. Vet. 19, 186–188 (2010).
Sacchi, A. B. V., Duarte, J. M. B., André, M. R. & Machado, R. Z. Prevalence and molecular characterization of Anaplasmataceae agents in free-ranging Brazilian marsh deer (Blastocerus dichotomus). Comp. Immunol. Microbiol. Infect. Dis. 32, 325–334 (2012).
Silveira, J. A., Rabelo, E. M. & Ribeiro, M. F. Molecular detection of tickborne pathogens of the family Anaplasmataceae in Brazilian Brown Brocket Deer (Mazama gouazoubira, Fischer, 1814) and Marsh Deer (Blastocerus dichotomus, Illiger, 1815). Transbound. Emerg. Dis. 59, 353–360 (2012).
Silveira, J. A. G. et al. Molecular detection and identification of hemoparasites in pampas deer (Ozotocerus bezoarticus Linnaeus, 1758) from the Pantanal Brazil. Ticks Tick Borne Dis. 4, 341–345 (2013).
Silveira, J. A. G., Rabelo, E. M. L., Lima, P. C. S., Chaves, B. N. & Ribeiro, M. F. B. Post-mortem hemoparasites detection in free-living Brazilian brown brocket deer (Mazama gouazoubira, Fischer 1814). Rev. Bras. Parasitol. Vet. 23, 206–215 (2014).
Mongruel, A. C. et al. Molecular characterization of Anaplasma sp. in free-living gray brockets (Mazama gouazoubira). Vector Borne Zoonotic Dis. 17, 165–171 (2016).
André, M. R. et al. Molecular detection of tick-borne bacterial agents in Brazilian and exotic captive carnivores. Ticks Tick Borne Dis. 3, 247–253 (2012).
Almeida, A. P., Souza, T. D., Marcili, A. & Labruna, M. B. Novel Ehrlichia and Hepatozoon agents infecting the Crab-Eating Fox (Cerdocyon thous) in Southeastern Brazil. J. Med. Entomol. 50, 640–646 (2013).
de Sousa, K. C. M. et al. Anaplasmataceae agents among wild mammals and ectoparasites in Brazil. Epidemiol. Infect. 145, 3424–3437 (2017).
André, M. R. et al. Molecular and detection of Ehrlichia spp. in endangered Brazilian wild captive felids. J. Wildl. Dis. 46, 1017–1023 (2010).
Widmer, C. E., Azevedo, F. C., Almeida, A. P., Ferreira, F. & Labruna, M. B. Tick-borne bacteria in free-living jaguars (Panthera onca) in Pantanal, Brazil. Vector Borne Zoonotic Dis. 11, 1001–1005 (2011).
Wolf, R. W. et al. Novel Babesia and Hepatozoon agents infecting non-volant small mammals in the Brazilian Pantanal, with the first record of the tick Ornithodoros guaporensis in Brazil. Ticks Tick Borne Dis. 7, 449–456 (2016).
Benevenute, J. L. et al. Assessment of a quantitative 5’ nuclease real-time polymerase chain reaction using groEL gene for Ehrlichia and Anaplasma species in rodents in Brazil. Ticks Tick Borne Dis. 8, 646–656 (2017).
Braga, M. S. C. O. et al. Molecular detection of Anaplasmataceae agents in Dasyprocta azarae in northeastern in Brasil. Rev. Bras. Parasitol. Vet. 27, 99–105 (2018).
Soares, H. S. et al. Ticks and rickettsial infection in the wildlife of two regions of the Brazilian Amazon. Exp. Appl. Acarol. 65, 125–140 (2017).
Lopes, M. G. et al. Ticks, rickettsial and erlichial infection in small mammals from Atlantic forest remnants in northeastern Brazil. Int. J. Parasit. Parasites Wildl. 7, 380–385 (2018).
Guimarães, A. et al. Detection of a putative novel genotype of Ehrlichia sp. from opossums (Didelphis aurita) from Brazil. Rev. Bras. Parasitol. Vet. 28, 140–144 (2019).
Machado, R. Z. et al. Migratory and carnivorous birds in Brazil: Reservoirs for Anaplasma and Ehrlichia species?. Vector Borne Zoonotic Dis. 12, 705–708 (2012).
Werther, K. et al. Arthropod-borne agents in wild Orinoco geese (Neochen jubata) in Brazil. Comp. Immunol. Microbiol. Infect. Dis. 55, 30–41 (2017).
Gardner, A. L. Order Pilosa in Mammal Species of the World (Johns Hopkins University Press, Baltimore, 2005).
Reis, N. R., Peracchi, A.L., Pedro, W. A., Lima, I. P. Mamíferos do Brasil (Eds, 2006).
Kluyber, D. et al. Ticks (Acari: Ixodidae) infesting armadillos (Cingulata: Dasypodidae) in the Pantanal wetland, Mato Grosso do Sul, Brazil. Syst. Appl. Acarol. 21, 1087–1092 (2016).
Guillemi, E. C. et al. Molecular identification of Anaplasma marginale in two autochthonous South American wild species revealed an identical new genotype and its phylogenetic relationship with those of bovines. Parasit. Vectors 9, 305 (2016).
Birkenheuer, A. J., Levy, M. G. & Breitschwerdt, E. B. Development and evaluation of a seminested PCR for detection and differentiation of Babesia gibsoni (Asian genotype) and B. canis DNA in canine blood samples. J. Clin. Microbiol. 41, 4172–4177 (2003).
Massung, R. F. et al. Nested PCR assay for detection of granulocytic ehrlichiae. J. Clin. Microbiol. 36, 1090–1095 (1998).
Drazenovich, N., Foley, J. & Brown, R. N. Use of real-time quantitative PCR targeting the msp2 protein gene to identify cryptic Anaplasma phagocytophilum infections in wildlife and domestic animals. Vector Borne Zoonotic Dis. 6, 83–90 (2006).
Carelli, G. et al. Detection and quantification of Anaplasma marginale DNA in blood samples of cattle by real-time PCR. Vet. Microbiol. 124, 107–114 (2007).
Rejmanek, D., Bradburd, G. & Foley, J. Molecular characterization reveals distinct genospecies of Anaplasma phagocytophilum from diverse North American hosts. J. Med. Microbiol. 61, 204 (2012).
Doyle, C. K. et al. Detection of medically important Ehrlichia by quantitative multicolor TaqMan real-time polymerase chain reaction of the dsb gene. J. Mol. Diagn. 7, 504–510 (2005).
Reller, M. & Dumler, J. Development and clinical validation of a multiplex real-time quantitative PCR assay for human infection by Anaplasma phagocytophilum and Ehrlichia chaffeensis. Trop. Med. Infect. Dis. 3, 14 (2018).
Müller, A. et al. “Candidatus Neoehrlichia chilensis” sp. Nov.: Molecular detection and characterization of a novel Anaplasmataceae in wild rodents from Valdivia, southern Chile. Transbound. Emerg. Dis. 65, 357–362 (2018).
Nion, V. L. et al. Potentially novel Ehrlichia species in horses, Nicaragua. Emerg. Infect. Dis. 21, 335 (2015).
Inayoshi, M., Naitou, H., Kawamori, F., Masuzawa, T. & Ohashi, N. Characterization of Ehrlichia species from Ixodes ovatus ticks at the foot of Mt. Fuji, Japan. Microbiol. Immunol. 48, 737–745 (2004).
Bustin, S. A. et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55, 611–622 (2009).
Aguiar, D. M., Saito, T. B., Hagiwara, M. K., Machado, R. Z. & Labruna, M. B. Serological diagnosis of canine monocytic ehrlichiosis with Brazilian antigen of Ehrlichia canis. Cien. Rural 37, 796–802 (2007).
Sanger, F., Nicklen, S. & Coulson, A. R. DNA sequencing with chain-terminating inhibitors. Proc. Nati. Acad. Sci. 74, 5463–5467 (1977).
Ewing, B., Hillier, L., Wendl, M. C. & Green, P. Base-calling of automated sequencer traces usingPhred. I. Accuracy assessment. Genome Res. 8, 175–185 (1998).
Ewing, B. & Green, P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 8, 186–194 (1998).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Benson, D. A. et al. GenBank. Nucleic Acids Res. 41, D36–D42 (2012).
Thompson, J. D., Higgins, D. G. & Gibson, T. J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680 (1994).
Hall, T. A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. In Nucleic acids symposium series, Vol. 41, No. 41, 95–98 (Information Retrieval Ltd., London, c1979–c2000 (1999).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 9, 772 (2012).
Miller, M. A., Pfeiffer, W., & Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the Gateway Computing Environments Workshop (GCE), 01–08 (2010).
Ronquist, F. & Huelsenbeck, J. P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574 (2003).
Stamatakis, A., Hoover, P. & Rougemont, J. A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 57, 758–771 (2008).
Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 39, 783–791 (1985).
Stöver, B. C. & Müller, K. F. TreeGraph 2: Combining and visualizing evidence from different phylogenetic analyses. BMC Bioinform. 11, 7 (2010).
Librado, P. & Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452 (2009).
Clement, M., Posada, D. & Crandall, K. TCS: A computer program to estimate gene genealogies. Mol. Ecol. 9, 1657–1660 (2000).
Huson, D. H. & Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267 (2005).
Leigh, J. W. & Bryant, D. Popart: full-feature software for haplotype network construction. Methods Ecol. Evol. 6, 1110–1116 (2015).
Fry, N. K., Warwick, S., Saunders, N. A. & Embley, T. M. The use of 16S ribosomal RNA analyses to investigate the phylogeny of the family Legionellaceae. Microbiology 137, 1215–1222 (1991).
Clarridge, J. E. Impact of 16S rRNA gene sequence analysis for identification of bacteria on clinical microbiology and infectious diseases. Clin. Microbiol. Rev. 17, 840–862 (2004).
Koh, F. X., Kho, K. L., Panchadcharam, C., Sitam, F. T. & Tay, S. T. Molecular detection of Anaplasma spp. in pangolins (Manis javanica) and wild boars (Sus scrofa) in Peninsular Malaysia. Vet. Parasitol. 227, 73–76 (2016).
Battilani, M., De Arcangeli, S., Balboni, A. & Dondi, F. Genetic diversity and molecular epidemiology of Anaplasma. Infect. Genet. Evol. 49, 195–211 (2017).
Cabezas-Cruz, A. et al. Ehrlichia minasensis sp. Nov., isolated from the tick Rhipicephalus microplus. Int. J. Syst. Evol. Microbiol. 66, 1426–1430 (2016).
Aguiar, D. M., Araujo, J. P., Nakazato, L., Bard, E. & Cabezas-Cruz, A. Complete genome sequence of an Ehrlichia minasensis strain isolated from cattle. Microbiol. Resour. Announc. 8, e00161-e219 (2019).
Aguiar, D. M. et al. A novel Ehrlichia genotype strain distinguished by the TRP36 gene naturally infects cattle in Brazil and causes clinical manifestations associated with ehrlichiosis. Ticks Tick Borne Dis. 5, 537–544 (2014).
Gajadhar, A. A., Lobanov, V., Scandrett, W. B., Campbell, J. & Al-Adhami, B. A novel Ehrlichia genotype detected in naturally infected cattle in North America. Vet. Parasitol. 173, 324–329 (2010).
Hailemariam, Z. et al. Molecular detection of tick-borne pathogens in cattle from Southwestern Ethiopia. PLoS ONE 12, e0188248 (2017).
Lobanov, V. A., Gajadhar, A. A., Al-Adhami, B. & Schwantje, H. M. Molecular study of free-ranging mule deer and white-tailed deer from British Columbia, Canada, for evidence of Anaplasma spp. and Ehrlichia spp.. Transbound. Emerg. Dis. 59, 233–243 (2012).
Cabezas-Cruz, A. et al. New species of Ehrlichia isolated from Rhipicephalus (Boophilus) microplus shows an ortholog of the E. canis major immunogenic glycoprotein gp36 with a new sequence of tandem repeats. Parasites Vectors 5, 291 (2012).
Iweriebor, B. C. et al. Genetic profiling for Anaplasma and Ehrlichia species in ticks collected in the Eastern Cape Province of South Africa. BMC Microbiol. 17, 45 (2017).
Thomson, K. et al. A new TaqMan method for the reliable diagnosis of Ehrlichia spp. in canine whole blood. Parasites Vectors 11, 350 (2018).
Rehman, A., Conraths, F. J., Sauter-Louis, C., Krücken, J. & Nijhof, A. M. Epidemiology of tick-borne pathogens in the semi-arid and the arid agro-ecological zones of Punjab province, Pakistan. Transbound. Emerg. Dis. 66, 526–536 (2019).
Carvalho, I. T. et al. Minimum infection rate of Ehrlichia minasensis in Rhipicephalus microplus and Amblyomma sculptum ticks in Brazil. Ticks Tick Borne Dis. 7, 849–852 (2016).
Cicculli, V. et al. First detection of Ehrlichia minasensis in Hyalomma marginatum ticks collected from cattle in Corsica, France. Vet. Med. Sci. 5, 243–248 (2019).
Li, J. et al. Emergence of a novel Ehrlichia minasensis strain, harboring the major immunogenic glycoprotein trp36 with unique tandem repeat and C-terminal region sequences, in Haemaphysalis hystricis ticks removed from free-ranging sheep in Hainan Province, China. Microorganisms 7, 369 (2019).
Groves, M. G., Dennis, G. L., Amyx, H. L. & Huxsoll, D. L. Transmission of Ehrlichia canis to dogs by ticks (Rhipicephalus sanguineus). Am. J. Vet. Res. 36, 937–940 (1975).
Moraes-Filho, J., Krawczak, F. S., Costa, F. B., Soares, J. F. & Labruna, M. B. Comparative evaluation of the vector competence of four South American populations of the Rhipicephalus sanguineus group for the bacterium Ehrlichia canis, the agent of canine monocytic ehrlichiosis. PLoS ONE 10, e0139386 (2015).
Johnson, E. M. et al. Experimental transmission of Ehrlichia canis (Rickettsiales: Ehrlichieae) by Dermacentor variabilis (Acari: Ixodidae). Vet. Parasitol. 74, 277–288 (1998).
Kocan, K. M., De La Fuente, J. & Cabezas-Cruz, A. The genus Anaplasma: New challenges after reclassification. Rev. Sci. Tech. 34, 577–586 (2015).
Marques, S., Barros-Battesti, D. M., Faccini, J. L. H. & Onofrio, V. C. Brazilian distribution of Amblyomma varium Koch, 1844 (Acari: Ixodidae), a common parasite of sloths (Mammalia: Xenarthra). Mem. Inst. Oswaldo Cruz 97, 1141–1146 (2002).
Soares, H. S. et al. Ticks and rickettsial infection in the wildlife of two regions of the Brazilian Amazon. Exp. Appl. Acarol. 65, 125–140 (2015).
Martins, T. F. et al. Diversity of ticks in the wildlife screening center of São Paulo city, Brazil. Cien. Rural 47, e20161052 (2017).
Muñoz-García, C. I. et al. Epidemiological study of ticks collected from the northern tamandua (Tamandua mexicana) and a literature review of ticks of Myrmecophagidae anteaters. Ticks Tick Borne Dis. 10, 1146–1156 (2019).
Acknowledgements
The authors would like to thank “Fundação de Amparo à Pesquisa do Estado de São Paulo” for the financial support (FAPESP Process number 2018/02753-0) and A.C.C.’s Master Scholarship (FAPESP Process number 2018/13838-6). MRA is a fellowship researcher of “Conselho Nacional de Desenvolvimento Científico e Tecnólogico” (CNPq Process number #302420/2017-7). We are also thankful to “Laboratório de Biologia Evolutiva e Conservação” tissue bank, Institute of Biology (IB), USP (Sao Paulo, SP, Brazil) and ICAS (Instituto de Conservação de Animais Silvestres—Projeto Bandeiras e Rodovias). We are also thankful to Renan Bressianini do Amaral for bioinformatics technical assistance.
Author information
Authors and Affiliations
Contributions
Conception and design: A.C.C. and M.R.A. Acquisition of data: A.C.C., J.G.V., M.H.A., D.R.Y., A.L.J.D., M.D.S., M.S.S., T.M.V.S., K.W. and M.M.G.T. Analysis and Interpretation of data: A.C.C., R.Z.M. and M.R.A. Drafting the article: A.C.C., J.G.V., M.H.A., D.R.Y., A.L.J.D., M.S., M.S.S., T.M.V.S., K.W., M.M.G.T., R.Z.M. and M.R.A.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Calchi, A.C., Vultão, J.G., Alves, M.H. et al. Ehrlichia spp. and Anaplasma spp. in Xenarthra mammals from Brazil, with evidence of novel ‘Candidatus Anaplasma spp.’. Sci Rep 10, 12615 (2020). https://doi.org/10.1038/s41598-020-69263-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-69263-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
